Gene Core – Kvantitativní a digitální PCR
Mezinárodní vědecké konsorcium pracuje na objasnění mechanismu ultra-vzácné Alexandrovy choroby
Alexandrova choroba je velmi vzácné neurodegenerativní onemocnění. Obvykle se projevuje v kojeneckém věku, i když jsou známy i jiné varianty objevující se v dospělosti. S cílem objasnění mechanismů Alexandrovy choroby vzniklo v nedávné době mezinárodní konsorcium, v němž má zastoupení i Česká republika prostřednictvím Laboratoře genové exprese (Biotechnologický ústav AV ČR v centru BIOCEV). Společně s partnery z různých evropských zemí si toto konsorcium klade za cíl pochopení mechanismu vzniku Alexandrovy choroby a s využitím unikátních modelů i nastínění možností pro budoucí léčbu.
Alexandrova choroba, kterou poprvé popsal v roce 1949 W. Stewart Alexander, je součástí skupiny vzácných progresivních neurologických onemocnění souhrnně nazývaných leukodystrofie. Leukodystrofie postihují převážně bílou hmotu nervového systému. Ta je tvořena velkým množstvím tzv. myelinu, což je základní protein obklopující výběžky neuronů a usnadňující jejich komunikaci. Abnormální vývoj či destrukce myelinu vede následně ke smrti neuronů a je příčinou zdravotních komplikací spojených s leukodystrofiemi.
Leukodystrofie jsou samy o sobě vzácné a postihují 1 až 2 lidi ze 100 tisíc. Alexandrova choroba tvoří pouhých několik procent z těchto případů. Nízká incidence dává Alexandrově chorobě status extrémně vzácného onemocnění, jehož skutečná prevalence je pouze odhadována. Jediná dokončená studie, která byla provedena v Japonsku a monitorovala četnost Alexandrovy choroby během pětiletého intervalu, stanovila její prevalenci na 1 případ na 2,7 milionu obyvatel. Celkově pak bylo na celém světě popsáno přibližně šest stovek případů od prvotního popisu Alexandrovy choroby na konci čtyřicátých let minulého století dodnes.
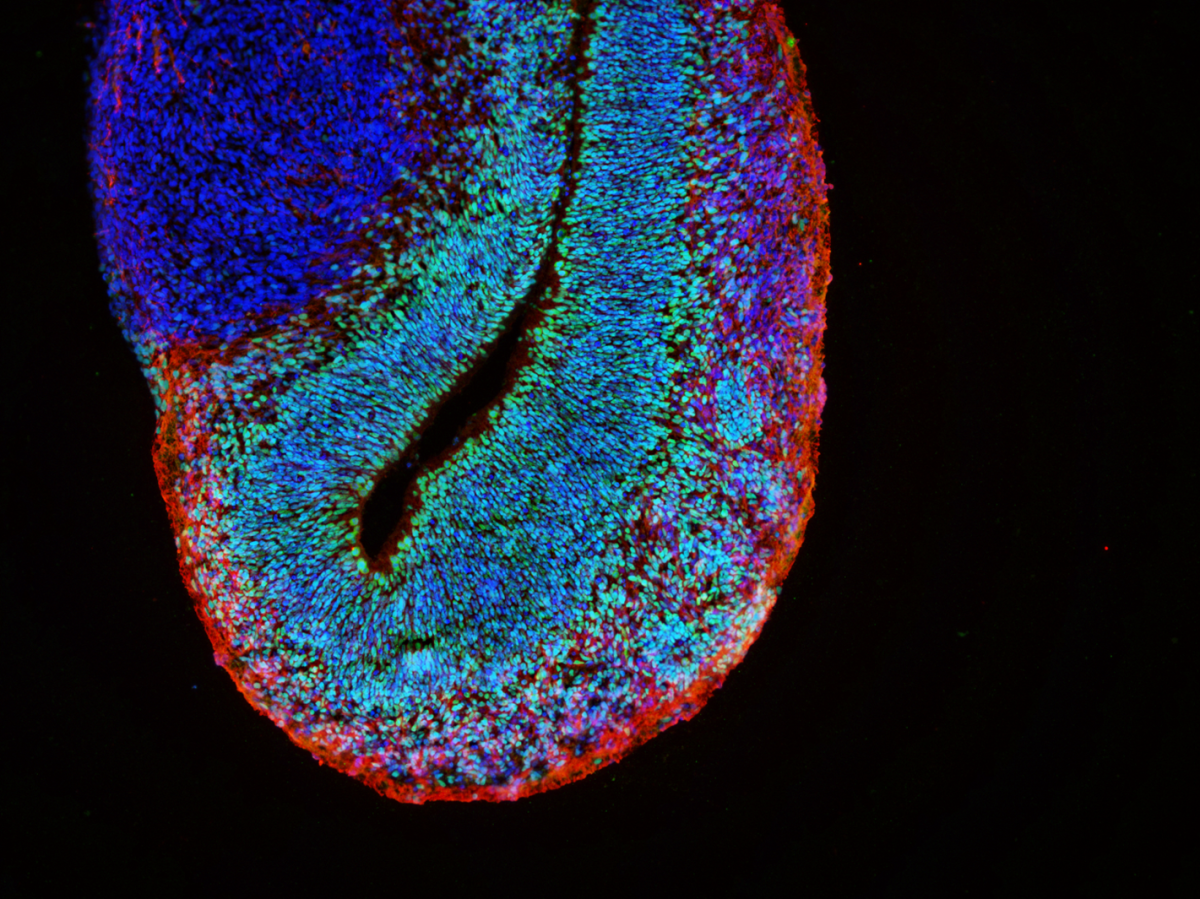
Diagnostika onemocnění byla dříve značně komplikovaná a zpravidla byla potvrzena až histologickým vyšetřením mozkové tkáně po smrti pacienta. Až koncem tisíciletí byly do klinické praxe zavedeny nové diagnostické metody. Magnetická rezonance (MRI) byla schopna vizualizovat úbytek bílé hmoty nervového systému a metody genetického testování pomohly přesně identifikovat změny v DNA způsobující Alexandrovu chorobu.
Alexandrova choroba je způsobena mutací genu GFAP, který kóduje gliální fibrilární kyselý protein. Tento protein je produkován zejména v astrocytech, buňkách, které podporují správnou funkci neuronů. Z tohoto důvodu je také Alexandrova choroba chápana jako primární onemocnění astrocytů, ačkoliv vliv dalších buněk nervové soustavy nebyl zcela vyloučen. Mutace genu GFAP způsobuje změnu vlastností vznikajícího proteinu. Jedním z důsledků je pak tvorba proteinových shluků, tzv. Rosenthalových vláken, uvnitř astrocytů. Tyto shluky se staly také prvním klíčovým identifikátorem při diagnostice Alexandrovy choroby v začátcích jejího studia. Změněná exprese genu GFAP pravděpodobně přispívá k aktivaci astrocytů a dalších buněk nervového systému, což vede k zánětu a konečné ztrátě bílé hmoty. Nicméně přesné mechanismy a zapojení jednotlivých buněčných typů zatím nejsou známy.
V současné době jsou popsány více než tři stovky mutací vedoucích ke vzniku Alexandrovy choroby. Přes 90 % těchto mutací je způsobeno pouhou jednou záměnou v sekvenci DNA, která je však dostatečná pro rozvoj onemocnění. Ve více než 80 % případů je zdrojem mutace mužská pohlavní buňka, při jejímž vývoji se tyto změny pravděpodobně odehrávají. Zda konkrétní typ mutace ovliňuje nástup a průběh onemocnění, není doposud známo, i když některé typy mutací se zdají být více asociované s časnějším nástupem onemocnění.
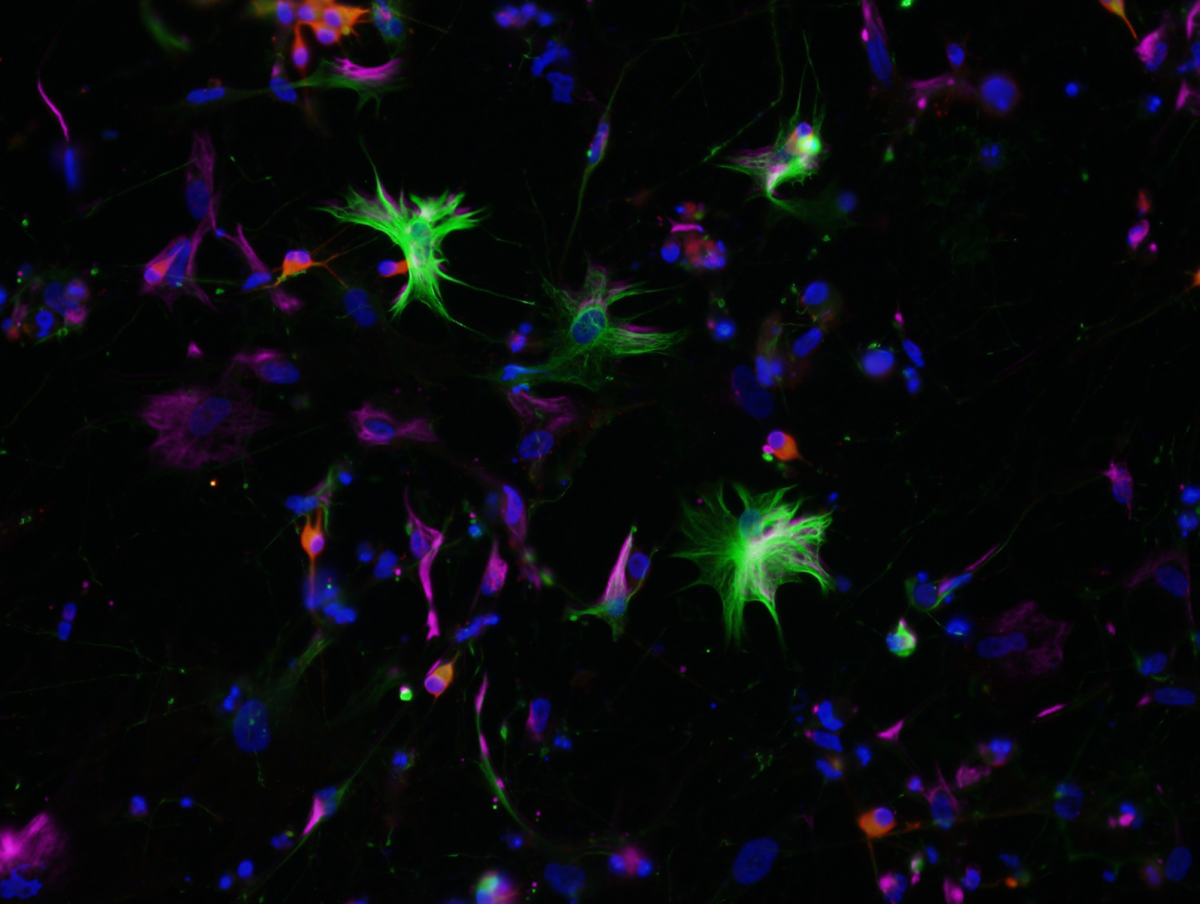
Onemocnění má mnoho klinických forem a projevuje se v různém věku, od narození po dospělost. Čím dříve se nemoc projeví, tím závažnější bývá její průběh. Nejčastější (více než 40 % případů) je novorozenecká forma, která se obvykle projeví během prvních dvou let života. Typicky při ní dochází k opoždění duševního i fyzického vývoje, abnormálnímu zvětšení hlavy, vyskytují se problémy s příjmem potravy a dítě často trpí záchvaty. Tito pacienti umírají obvykle během prvních několika let života. Mladistvá forma Alexandrovy choroby je méně častá a obvykle začíná ve věku od 4 do 15 let. Tyto děti mohou trpět nadměrným zvracením, mít potíže s polykáním a mluvením, špatnou koordinaci a ztrátu kontroly motoriky. Přežití je značně variabilní, vzácně se dožívají mladší dospělosti. Jako poslední jsou popsány i formy Alexandrovy choroby s nástupem v dospělosti. Část těchto případů však může unikat statistikám kvůli příznakům klinicky zaměňovaným s roztroušenou sklerózou nebo Parkinsonovou chorobou.
Bohužel dosud neexistuje žádná úspěšná terapie Alexandrovy choroby a léčba je převážně zaměřena na mírnění projevů onemocnění. Spektrum podpůrné léčby zahrnuje kontrolu záchvatů pomocí léků, odvod mozkové tekutiny, výživovou sondu, podání medikamentů na kontrolu refluxu a zvracení apod. Jistým příslibem do budoucnosti může být vývoj nových léčiv cílených na snížení exprese mutantního proteinu GFAP či oprava zmutované sekvence genu pomocí metod genové terapie. Další pokrok by mohlo přinést rovněž lepší pochopení mechanismu choroby, které by umožnilo nejenom cílený design nových léčiv, ale potenciálně i využití stávajících medikamentů.
S cílem objasnění mechanismů Alexandrovy choroby vzniklo v nedávné době i mezinárodní konsorcium, v němž má zastoupení i Česká republika prostřednictvím Laboratoře genové exprese sídlící v novém Biotechnologickém a biomedicínském centru Akademie věd a Univerzity Karlovy (BIOCEV) na okraji Prahy. Společně s partnery z různých evropských zemí si toto konsorcium klade za cíl pochopení mechanismu vzniku Alexandrovy choroby a s využitím unikátních modelů i nastínění možností pro budoucí léčbu. Projekt je součástí Evropského programu na výzkum vzácných onemocnění (EJP RD) financovaného v rámci Iniciativy na výzkum v oblasti vzácných onemocnění ERA-Net.
Pro budoucí generace pacientů mohou podobné projekty představovat kýžený zlom v léčbě této fatální neurodegenerativní choroby. Pro současnou generaci je nadějí časná diagnostika a podpůrná léčba. Diagnózu lze stanovit na základě klinických znaků a zobrazovacích metod a její potvrzení je provedeno pomocí genetického testování.
Autoři článku: Lukáš Valihrach a Zuzana Benešová (Laboratoř genové exprese, BTU AV ČR, BIOCEV)