What can be seen without labelling?
Fluorescence microscopy is a popular and powerful tool mainly due to its specificity and high signal-to-noise ratio, which is largely due to the low auto-fluorescence background of most of the studied samples. What happens when we insert an unlabelled sample into a fluorescence microscope? Will we see anything at all? And why should we actually work with unlabelled samples?
The reason for working with unlabelled samples is not only laziness or time and economic requirements, but especially the desire not to modify or damage the sample in any way. Fluorescent dyes, whether added to a sample in the form of probes penetrating the sample and tagging specific organelles, or in the form of genetically engineered fluorescent proteins, are not a natural component of cells and can modify cellular properties and behaviour. For each probe, it is necessary to test what concentrations begin to cause cell modifications, ultimately leading to its death. Each genetically modified fluorescently labelled protein should be checked to ensure that it has retained its original function and that its amount in the cell corresponds to normal physiological conditions. Thus, imaging methods not relying on labelling the samples have undeniable advantages. But what can we actually image without labelling?
With a touch of humor, it is sometimes said that auto-fluorescence signal is only a matter of the applied excitation laser power . However, too much light is detrimental to cells, especially in the UV spectral region, so it is vital to limit excitation power to cell-tolerant values. Popular targets for auto-fluorescence imaging are the coenzymes NAD(P)H and FAD, which are actively involved in cell metabolism. The ratio of oxidized and reduced states of these metabolites together with their total concentrations is particularly suitable for documenting the metabolic activity of cells. Measuring the metabolic activity, among other things, makes it possible to distinguish healthy tissues from cancerous growths. So how can metabolic activity be displayed?
Coincidentally, the reduced forms of the coenzymes NAD(P)H, and the oxidized form of FAD are weakly fluorescent. Although their luminosity is an order of magnitude worse than that of the fluorophores used for specific labelling, they replace quality with quantity, so the signal from the entire cell becomes strong enough for analysis. A certain disadvantage, especially for imaging of NAD(P)H fluorescence, is the need to use UV excitation, specifically around 370 nm. As stated above, living cells really do not like UV light, plus, most confocal microscopes lack a suitable laser and optimized optics for working with such short wavelengths. How can this be dealt with?
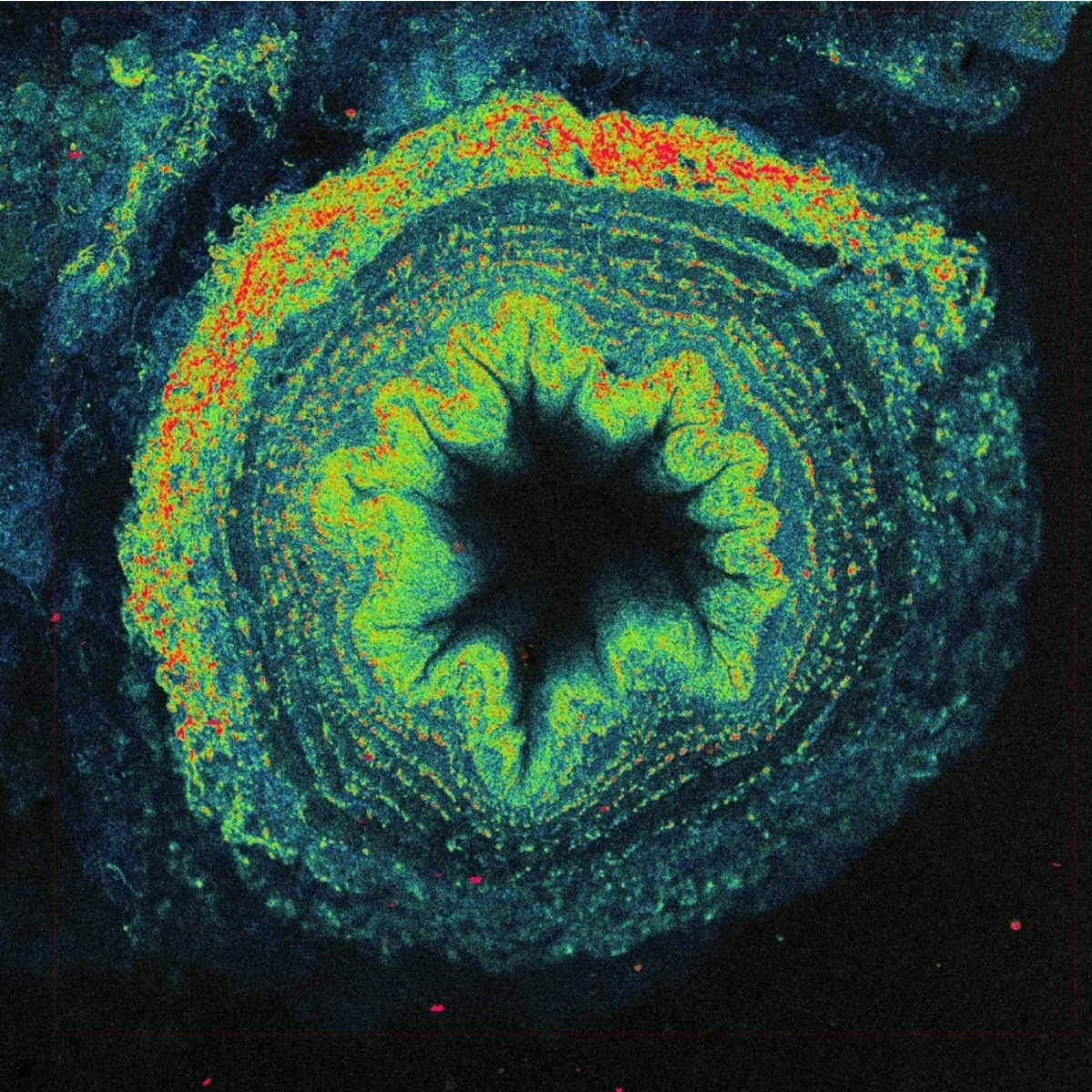
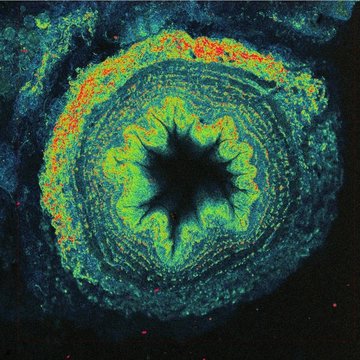
The solution is to use two photons to excite fluorescence instead of just one. One UV photon at 370 nm has the same energy as two near-infrared photons at 740 nm. Two-photon excitation with the near-infrared light has other advantages - thanks to less scattering and absorption, light penetrates better into the depth of biological samples and excitation is achieved only in a small volume, which automatically allows 3D imaging. However, simultaneous absorption of two photons is a very unlikely phenomenon, in fact several tens of orders of magnitude less probable than conventional single photon absorption. No wonder the technology needed to use two-photon excitation for imaging was not developed until the 1990s. We need very strong and short (around one hundred femtoseconds) laser flashes with a high repetition rate (tens of megahertz), perfect durable optics, fast scanner, and sensitive detectors.
Additionally, powerful lasers employed in two-photon fluorescence microscopy make it possible to utilize generation principles other than fluorescence for imaging. A popular method is second harmonic generation (SHG), where due to the local asymmetry of the arrangement of macromolecules, typically collagen or myosin fibres in animal cells, or starch or cellulose in plants, light is generated at exactly half the wavelength of the excitation used. For example, when using infrared excitation with 900 nm wavelength, blue light with a wavelength of 450 nm is produced, which gives a slightly different image than auto-fluorescence. Imaging native tissue structures, including muscle, is useful in the study of many degenerative diseases.
Another possibility is to use third harmonic generation (THG), where the signal is generated at optical interfaces, such as microdroplets, thanks to a combination of three photons to generate one-third of a wavelength. Here, it is necessary to use wavelengths above 1200 nm so that the generated signal is in the visible range and detectable. An interesting application is the imaging of red blood cells, which makes it possible to visualize small capillaries in tissues, including blood flow velocity.
The list of alternatives to fluorescence does not end there. An attractive source of sample information is the Raman scattering of light, because the Raman spectrum is highly specific and potentially allows for the identification of native molecules through their vibrational levels. Unfortunately, the Raman signal is very weak, so direct imaging is not very practical. (On[PD1] the other hand, if the Raman signal was too strong, it would interfere with fluorescence imaging because they spectrally overlap.) However, imaging based on vibrational properties is possible utilizing Coherent anti-Stokes Raman scattering (CARS), which uses two overlapping infrared beams to synchronize the transition between the selected vibrational states, thereby significantly amplifying the scattered signal. Lipid microdroplets are most often and most easily imaged due to their high concentration of CH2 groups.
It is obvious that even without labelling you can see quite a lot if you have the appropriate instrumentation. Microscopes equipped with powerful infrared lasers can be found at the Institute of Physiology, the Academy of Sciences of the Czech Republic, and at BIOCEV.
The mentioned procedures, techniques, and equipment are operated by the following workplaces: The Institute of Physiology, The Academy of Sciences of the Czech Republic - Bioimaging Facility, Charles University (BIOCEV) - Imaging Methods, Brno University of Technology (CEITEC) - Experimental Biophotonics.
Author: Aleš Benda, Head of Imaging Methods Core Facility