From organelles to molecules: super-resolution microscopy
Even the best optical lens does not allow us to directly see details of cells that are smaller than approximately 200-300 nm. Light diffraction shatters the signal from any small object so much that if two objects are closer than about half the wavelength of the visible light used, their diffraction patterns almost completely overlap and the observer is unable to tell if they are seeing just one object or multiple objects. It was not until the turn of the millennium that this physical barrier was circumvented through super-resolution microscopy. This discovery was awarded with the Nobel Prize in Chemistry in 2014.
The operating principles of super-resolution microscopy allow us to understand the connection between chemistry and microscopy. The foundation lies in a fluorescent molecule and our ability to switch it back and forth between a fluorescent to non-fluorescent state. One could say this is molecular manipulation, which is made possible by the special chemical structure and properties of custom-synthesized organic molecules or genetically modified fluorescent proteins. This basic principle is used in dozens of different super-resolution procedures, each of which is suitable for a different type of sample. Three microscopic methods using structured illumination (SIM), stimulated emission depletion (STED) or single-molecule localization (SMLM) have become some of the most widely used commercial applications.
In Structured Illumination Microscopy (SIM), instead of employing traditional homogeneous illumination of the sample, illumination using a regular, finest possible pattern is used. By moving and rotating the excitation pattern, different parts of the sample are alternately displayed, and by analysing the thus obtained image sequence, a resulting image with up to twice the resolution of a conventional camera image can be obtained. SIM is advantageous because of its speed, applicability for almost any type of fluorescent labelling, multicolour, and medium dose of light used, thus making it reasonably compatible for observing living cells.
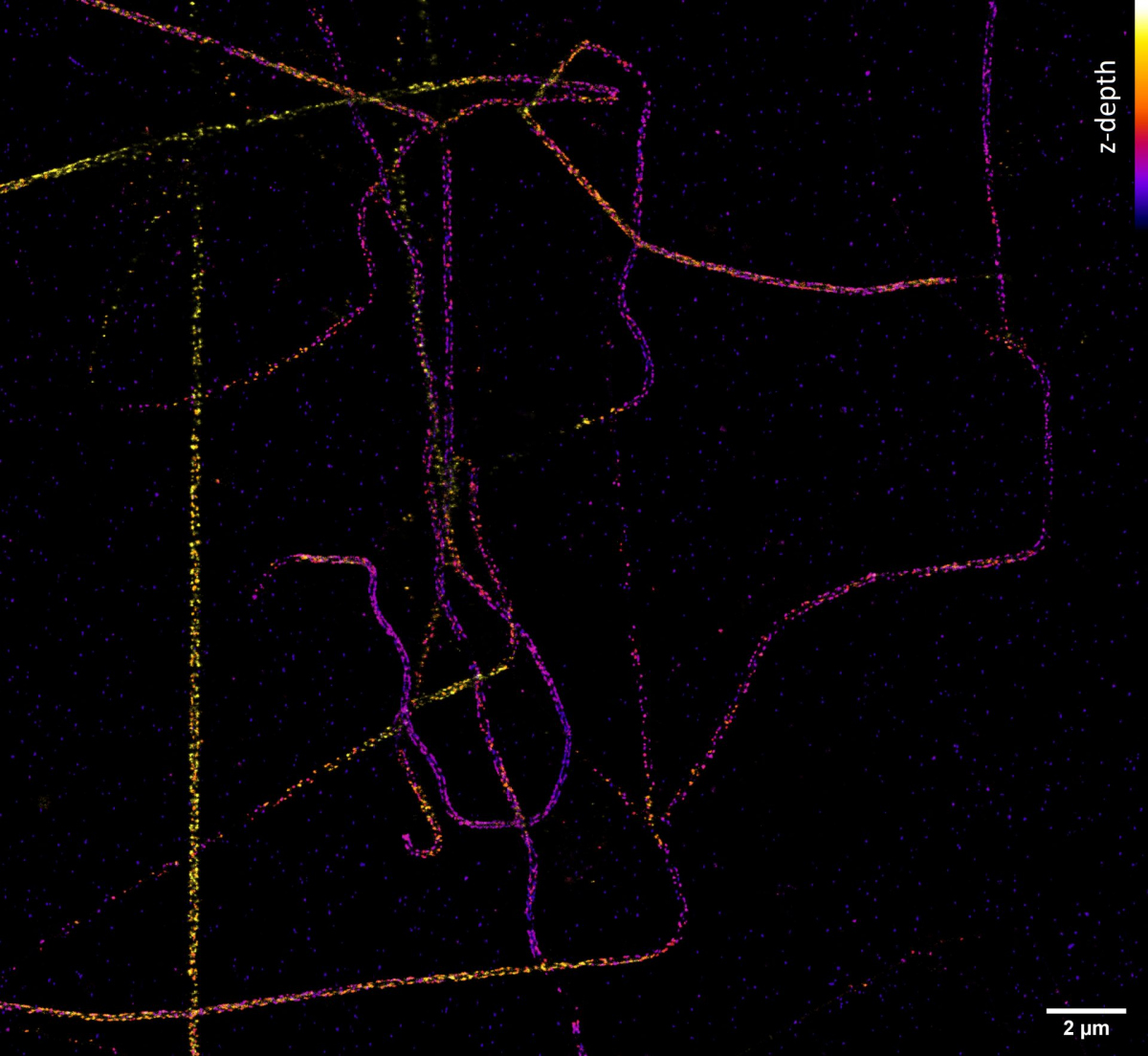
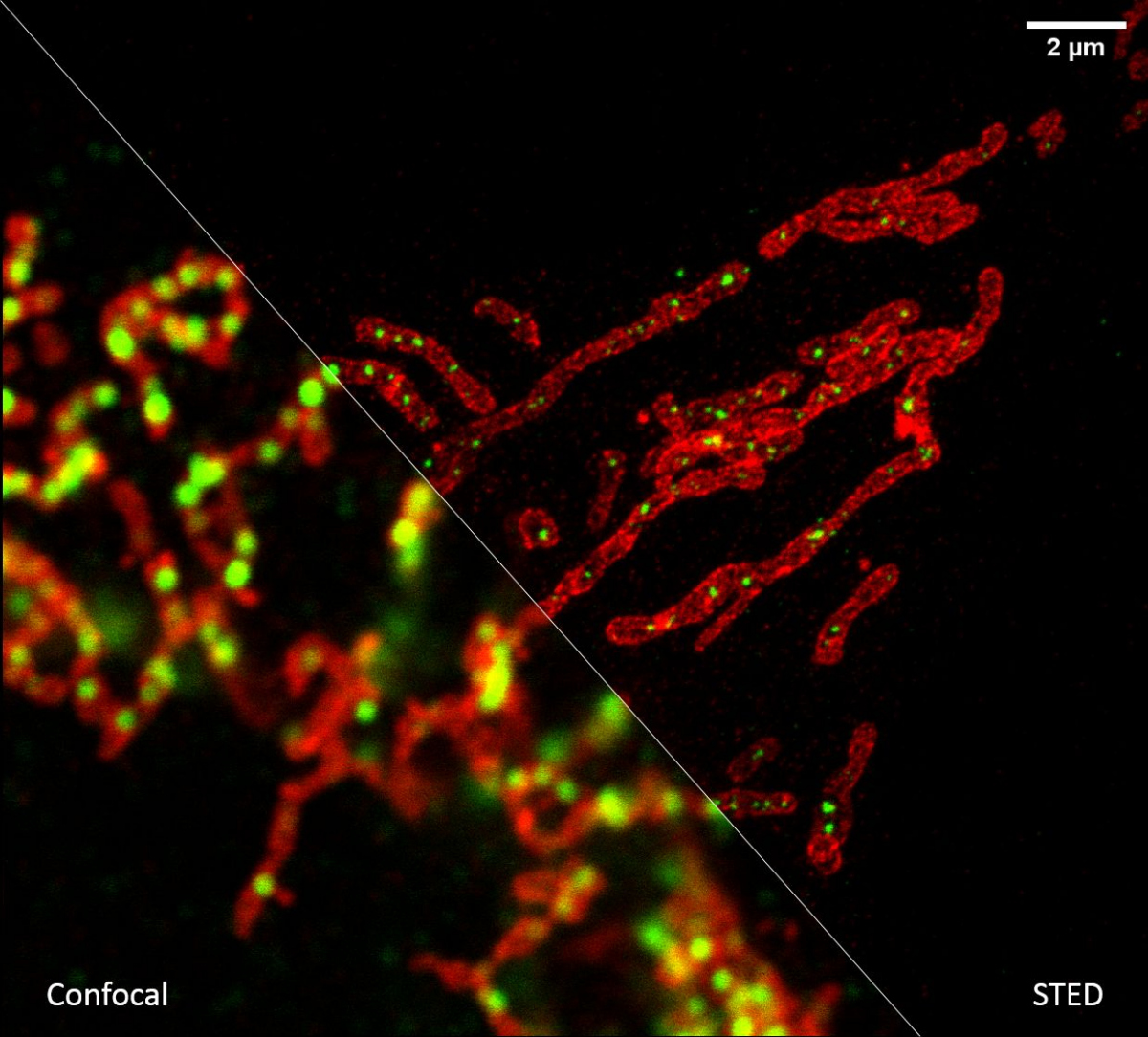
If we need to look at cell structures smaller than 100 nm, we can use Stimulated Emission Depletion (STED) microscopy. This method is based on a classical confocal laser scanning microscope and the use of a very powerful pulsed laser with a colour corresponding to the red part of the emission spectrum of the colour used. This laser is able to stimulate emissions in the excited molecules and thus switch them back to a ground state, thus preventing their spontaneous fluorescence. High resolution is achieved by overlapping the depletion laser (775 nm) with excitation lasers (e.g. 561 nm and 640 nm) with one important difference - it has no maximum in the centre, but zero intensity. Thus, effective stimulated emission occurs mainly at the edges of the original confocal volume and the fluorescence signal is thus detected with only a significantly reduced volume. This achieves a resolution of up to 30 nm in the scanned plane for 2D adjustment, or isotropic 120 nm in all axes for 3D adjustment. It is a purely optical phenomenon, without the need to resort to using any mathematical tricks with data, and thus with a minimal risk of artefacts. The method works great for two colours, where thanks to the shared depletion, two-colour images are always perfectly overlapped. However, a powerful depletion laser causes rapid photo-destruction, and the method thus requires very stable organic colours. STED is not very compatible with fluorescent proteins in living cells or with three- and multicolour scanning.
But how do we achieve a resolution of tens of nanometres and better, which we need, for example, to determine the arrangement of individual proteins in large protein complexes? Quite easily. If we know that we can capture only one single molecule at a given place and time, then although the camera shows us a blurred diffraction pattern, the fluorescent molecule is located exactly in the middle. The accuracy of the localization of a molecule then depends on its luminosity and the quality of the microscope - and it reaches nanometres! The Single Molecule Localization Microscopy (SMLM) method consists of randomly switching molecules from a fluorescent to a dark state so that a statistically maximum of one is always bright at a given place and time. Thus, if we cannot distinguish the emission of individual molecules in space due to diffraction, we can distinguish it in time. Commercial SMLM microscopes can obtain a map of localizations with an accuracy of typically around 10-20 nm in the xy plane of sharpness and 30-50 nm in the z-axis. However, the resolution of the image is usually not limited by localization accuracy, but by the quality of the sample used, especially how densely and with how well the sample was labelled.
Not sure which super-resolution method is right for your sample? Not to worry, you can try all three within the Czech-BioImaging infrastructure!
Author: Aleš Benda, Head of Imaging Methods Core Facility